{kind=link}
Parkinsonism-plus syndromes
Parkinsonism-plus syndromes หรือ Atypical parkinsonian syndromes หรือที่เรียกกันว่า โรคพาร์กินสันเทียม หมายถึงกลุ่มโรคที่มีอาการพาร์กินโซนิซึม คล้ายโรคพาร์กินสัน หากแต่ว่าการดำเนินโรค หรือลักษณะทางพยาธิวิทยาต่างจากโรคพาร์กินสัน คำว่า "-plus" นั้นมีความสำคัญ ซึ่งบ่งถึงอาการเพิ่มเติมที่นอกเหนือจากอาการพาร์กินโซนิซึม ซึ่งมักจะเป็นสิ่งที่แพทย์ควรสังเกต ตรวจหา และใช้เป็นข้อสังเกตว่าผู้ป่วยที่มีลักษณะอาการเพิ่มเติม เหล่านั้นอาจไม่ใช่ผู้ป่วยพาร์กินสันโดยแท้จริง หรือเป็นโรคพาร์กินสันเทียม.
เนื่องจากโรคพาร์กินสันเป็นโรคที่เริ่มด้านใดด้านหนึ่ง (asymmetric onset) และส่วนมากเริ่มด้วยอาการสั่น ประกอบกับอาการพาร์กินโซนิซึมอื่นๆ ดังที่ได้กล่าวไว้ข้างต้น ร่วมกับการตอบสนองที่ดีต่อยาลีโวโดปา และมีปัญหาในเรื่องของการตอบสนองต่อยาไม่สม่ำเสมอได้. เมื่อได้รับยาลีโวโดปาไประยะหนึ่ง ปัญหาในเรื่องของการเดิน การพูด และ cognitive impairment มักเกิดขึ้นในภายหลัง ดังนั้นอาการที่นอกเหนือจากนี้ อาจเป็นอาการชี้แนะสำหรับกลุ่มโรคพาร์กินสันเทียม.
Parkinsonism-plus syndromes ประกอบด้วยหลายโรค และมีชื่อที่ค่อนข้างจะจำยากและมี อาจชื่อที่เปลี่ยนไปจากสมัยก่อน. แต่สิ่งที่สำคัญที่แพทย์ควรเข้าใจ คือชื่อเหล่านั้นมักจะบ่งบอกถึงอาการ หลักที่เพิ่มขึ้นนอกเหนือจากอาการพาร์กินโซนิซึม ยกตัวอย่าง เช่น โรค Progressive supranuclear palsy (PSP) มีอาการเด่นคือ ปัญหาในเรื่องของการกลอกตาโดยเฉพาะในส่วนของการมองขึ้นลง. โรค Multiple system atrophy (MSA) เป็นโรคที่มีพยาธิสภาพที่เกิดในหลายๆ ระบบ ดังเช่นระบบประสาทอัตโนมัติ (autonomic nervous system).
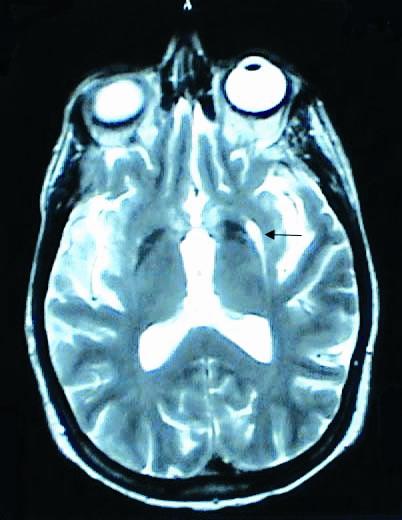
ภาพที่ 7. ภาพ MRI ของผู้ป่วย multiple system atrophy.
1. Progressive supranuclear palsy (PSP)
ดังชื่อของโรค ลักษณะอาการเด่นของโรค PSP ที่นอกเหนือจากอาการพาร์กินโซนิซึม คือปัญหาในการกลอกตา โดยเฉพาะในเรื่องของการมองขึ้นลง. PSP แต่เดิมชื่อว่า Steele-Richardson-Olzewski syndrome ถึงแม้ว่าปัญหาในเรื่องของการกลอกตาจะเป็นอาการเด่น แต่โดยส่วนใหญ่ผู้ป่วย PSP มีปัญหาในเรื่องการเดิน และหกล้มเป็นอาการแรก ซึ่งต่างจากผู้ป่วยโรคพาร์กินสันที่ปัญหาในการเดินและหกล้ม มักมาภายหลังจากมีอาการแล้วเป็นเวลา 2 ปีเป็นต้นไป. นอกจากนี้ อาการระยะแรกเริ่มในผู้ป่วย PSP จะเป็นในเรื่องของการเคลื่อนไหวช้า โดยทั่วไปอาการสั่นมีน้อยหรือไม่เกิดขึ้นเลย ซึ่งเมื่อรวมกับปัญหาในเรื่องของการเดินแล้ว อาจทำให้ผู้ป่วย PSP ในระยะแรกได้รับการวินิจฉัยว่าเป็นโรคอื่นๆ ดังเช่น โรคของประสาทหู โรคของไขสันหลัง เป็นต้น.
ปัญหาของการกลอกตา (eye movement disorders) เป็นอาการที่เกิดขึ้นภายหลัง แต่เมื่อเกิดแล้ว สามารถช่วยให้แพทย์วินิจฉัยและนึกถึงโรค PSP มากขึ้น. โดยทั่วไปแพทย์มักเข้าใจว่าผู้ป่วย PSP จะมีปัญหาเฉพาะในเรื่องของการมองขึ้นลง (vertical gaze palsy) แต่โดยแท้จริงแล้ว ปัญหาในการกลอกตา ที่เริ่มเป็นปัญหาแรกคือ slow saccades. เพราะฉะนั้น การตรวจการเคลื่อนไหวของตาที่สำคัญในผู้ป่วย PSP คือการตรวจระบบ saccades ทั้งในแนวตั้ง และแนวนอน. ผู้ป่วย PSP ในระยะแรกมักมี slow saccades ก่อนที่จะมีการมองขึ้นลง ซึ่งจะเป็นในเรื่องของการมองลงก่อน (downward gaze palsy) ก่่อนที่จะมีปัญหาในเรื่องของการมองขึ้น (upward gaze palsy). ในระยะท้ายผู้ป่วย PSP จะมีปัญหาในการมองในด้านข้างด้วย (horizontal gaze palsy) ส่งผลให้เกิด complete หรือ almost complete ophthalmoplegia ได้.9
หลังจากตรวจได้ว่าผู้ป่วยมีปัญหาของการมองในแนวตั้งแล้ว การตรวจที่สำคัญในขั้นต่อไป คือการตรวจ Vestibulo-ocular reflex (VOR) ทั้งในแนวนอนและแนวตั้ง ถ้าการทำ VOR ส่งผลให้ผู้ป่วยสามารถกลอกตาไปมาได้ จะบ่งถึงรอยโรคที่อยู่เหนือจาก nucleus (ไม่ได้อยู่ใน VOR pathway) หรือที่เรียกว่า supranuclear นั่นเอง ซึ่งถ้าเกิด ร่วมกับอาการพาร์กินโซนิซึม และปัญหาโดยเฉพาะในการเดินด้วยแล้ว จะทำโอกาสที่ผู้ป่วยนั้นเป็น PSP จะมีมากขึ้น.
อาการอื่นๆ ในผู้ป่วย PSP ที่นอกเหนือจากอาการข้างต้น จะเป็นในเรื่องของการพูดลำบาก (dysarthria) อาการดีสโตเนียที่คอ โดยเฉพาะเมื่อคอถูกดึงไปข้างหลัง (retrocollis). อาการของผู้ป่วย PSP มักสังเกตได้จากปัญหาในการขึ้นลงบันได เนื่องจากปัญหาของการทรงตัวซึ่งมักจะเอนตัวไปข้างหลัง คอแข็งทำให้แหงนคอขึ้นลงได้ยาก และปัญหาในการมองขึ้นลง. ลักษณะอาการต่างๆ ที่ได้กล่าวไปนี้ เป็นอาการหลักที่ใช้ในการวินิจฉัย PSP ตาม NINDS-SPSP diagnostic criteria.10
การวินิจฉัยโรค PSP อาศัยลักษณะอาการดังกล่าวข้างต้นตาม diagnostic criteria. การตรวจทางรังสีวิทยาหรือห้องปฏิบัติการจะเป็นการตรวจเสริมเพื่อช่วยยืนยันหรือเพื่อช่วยในการแยกโรค. โดยทั่วไปการตรวจด้วยเอกซเรย์คลื่นแม่เหล็ก (MRI) หรือทางรังสีวิทยา (CT) ทางระบบประสาทมักไม่พบความปกติที่เฉพาะต่อโรค. PSP ในผู้ป่วยส่วนน้อยอาจพบการแคบลง (anteroposterior diameter) ของส่วน midbrain tectum หรือการฝ่อลงของ colliculi ร่วมกับ third ventricle ที่กว้างใหญ่ขึ้น ซึ่งถ้าพบลักษณะดังกล่าว ก็จะช่วยชี้แนะหรือสนับสนุนถ้า ผู้ป่วยนั้นมีอาการของโรค PSP แต่ถ้าไม่พบลักษณะดังกล่าว ก็ไม่ใช่เป็นตัวตัดสินว่าผู้ป่วยนั้นไม่ได้เป็นโรค PSP. การตรวจทางน้ำไขสันหลัง อาจพบการเพิ่มขึ้นของโปรตีน Tau ซึ่งการตรวจดังกล่าว ยังเป็นในลักษณะของการวิจัย และไม่มีแพร่หลายสำหรับเวชปฏิบัติ.
ลักษณะทางพยาธิวิทยาของโรค PSP มีความแตกต่างจากโรคพาร์กินสัน โดยที่มีการเสื่อมของระบบประสาทในส่วนของ dopamine และ acetylcholine ที่เด่น. นอกเหนือจากส่วนของ substantia nigra และการพบ Neurofibrillary tangles (NFT) ซึ่งประกอบด้วยโปรตีน Tau (โปรตีน Tau เป็นองค์ประกอบสำคัญ ของ microtubule ที่มีหน้าที่เกี่ยวกับระบบนำส่งของเซลล์นั้นๆ) ที่มีลักษณะที่ผิดปกติ (abnormally phosphorylated tau ที่ไม่จับเป็นคู่) การตรวจทางพยาธิวิทยาที่อาศัยการย้อมชิ้นเนื้อด้วย anti-tau antibody ช่วยในการตรวจพบ NFTs ที่ผิดปกติ และกระจายในสมองส่วนต่างๆ ดังเช่นในส่วนของ cerebral cortex ส่วนของ nigrostriatal pathway ในส่วนของ pontomesencephalic region และในส่วนของ hindbrain ซึ่งลักษณะทางพยาธิวิทยาที่กระจายไปในส่วนต่างๆ เหล่านี้ส่งผลให้ผู้ป่วยมีอาการต่างๆ ไม่ว่าจะเป็นในเรื่องของการเดินที่เด่น การพูดที่ลำบาก และอื่นๆ ที่นอกเหนือ และต่างจากผู้ป่วยโรคพาร์กินสัน.
2. Multiple system atrophy (MSA)
MSA เป็นชื่อโรครวมและใหม่ ประกอบด้วยโรคย่อยที่มีลักษณะเด่นที่สำคัญที่แตกต่างกันไป แต่มีลักษณะร่วมที่เหมือนกันคือ เป็นโรคที่มีอาการร่วมของหลายๆ ระบบนอกเหนือจากอาการพาร์กินโซนิซึม (ดังชื่อ multiple system)
2.1 Multiple system atrophy-parkinsonism subtype (MSA-P, Striatnigral degeneration)
2.2 Multiple system atrophy-autonomic subtype (MSA-A, Shy-Drager syndrome)
2.3 Multiple system atrophy-cerebellar subtype (MSA-C, Olivopontocerebellar atrophy)
ดังนั้น ผู้ป่วย MSA จึงสามารถมีอาการที่แตกต่างกันไปนอกเหนือจากอาการพาร์กินโซนิซึม ซึ่งขึ้นอยู่กับว่า ผู้ป่วยท่านนั้นมีอาการเสื่อมของระบบใดที่เด่น ดังเช่น ถ้ามีอาการในส่วนของความดันโลหิตที่ควบคุมลำบาก ร่วมกับปัญหาในเรื่องของการขับถ่าย ซึ่งทั้ง 2 อาการเป็นระบบประสาทอัตโนมัติ ก็จะเป็นลักษณะของโรค MSA-A เป็นต้น. ในบางครั้งอาการต่างๆ เหล่านี้ไม่ได้เกิดขึ้นพร้อมๆ กัน ต้องอาศัยเวลาก่อนที่อาการต่างๆ จะค่อยๆ แสดงชัดเจนขึ้นในผู้ป่วย โดยที่ผู้ป่วยเพียงร้อยละ 28 เท่านั้นที่จะมีอาการครบทั้งอาการพาร์กินโซนิซึม อาการทางระบบประสาทอัตโนมัติ และอาการของสมองส่วนหลัง หรือ cerebellum. นอกจากนี้ ผู้ป่วย MSA สามารถมีอาการอื่นๆ ที่นอกเหนือจากดังที่ได้กล่าวไว้ข้างต้นได้ ดังเช่น pyramidal signs หรือ peripheral neuropathy เป็นต้น.
อาการพาร์กินโซนิซึมในผู้ป่วย MSA โดยส่วนใหญ่จะเป็นอาการเคลื่อนไหวช้า และเกร็ง ร่วมกับปัญหาในเรื่องของการเดินที่เกิดพร้อมๆ กันทั้ง 2 ข้างของร่างกาย อาการสั่นจะน้อย ต่างจากผู้ป่วยพาร์กินสันที่อาการจะเริ่มด้านใดด้านหนึ่ง และมีอาการสั่นเป็นอาการเด่น. อาการพาร์กินโซนิซึมจะเกิดใน ผู้ป่วยพาร์กินโซนิซึมทุกๆ คน ไม่ว่าจะเป็น MSA-P, MSA-A หรือ MSA-C หากแต่ว่ามีลักษณะที่เด่นชัดแตกต่างกันไป.
อาการทางระบบประสาทอัตโนมัติ สามารถเกิดได้ทั้งในผู้ป่วย MSA และผู้ป่วยพาร์กินสัน ซึ่งมักจะมีอาการที่ชัดกว่า รุนแรงกว่าในผู้ป่วย MSA inspiratory stirdor เป็นอาการที่เฉพาะ ถ้าเกิดขึ้นจะช่วยในการวินิจฉัย MSA ในผู้ป่วยรายนั้น.11 ส่วน orthostatic hypotension (หมายถึง ความดันที่ลดลงมากกว่า 20 มม.ปรอท ของ systolic blood pressure เมื่อผู้ป่วยลุกขึ้นยืนจากท่านั่ง) เป็นอาการที่พบบ่อยที่สุดในผู้ป่วย MSA ซึ่งอาการที่แสดงออกเนื่องจาก orthostatic hypotension ได้แก่ อาการมึนงง เป็นลม (syncope) เหนื่อย เมื่อยล้า เป็นต้น ซึ่งผู้ป่วยเป็นส่วนน้อยที่สามารถบอกอาการดังกล่าวว่าเกี่ยวข้องกับการเปลี่ยนท่าท่าง นอกเหนือจากปัญหาในเรื่องของการควบคุมความดันโลหิต อาการทางระบบขับถ่าย ก็พบได้บ่อย ดังเช่น ปัญหาในการควบคุมปัสสาวะ ซึ่งมักเป็นผลร่วมมาจาก detrusor hyperreflexia, urethral sphincter weakness และ failure of detrusor contraction อาการท้องผูก ปัญหาในการเสื่อมสมรรถนะทางเพศ อวัยวะเพศไม่แข็งตัวในผู้ป่วยชาย ปัญหาในเรื่องของการกลืนสามารถพบได้บ่อย เช่นเดียวกัน ปัญหาทางการหายใจ โดยเฉพาะ inspiratory stirdor ที่เกิดเนื่องมาจาก bilateral abductor vocal fold paresis สามารถส่งผลอันตรายถึงชีวิตในผู้ป่วย MSA ได้.12
อาการของสมองส่วนหลัง (cerebellar dysfunction) ในผู้ป่วย MSA โดยส่วนใหญ่จะเป็นปัญหาของการเดิน (gait ataxia) ร่วมกับปัญหาในการพูด (dysarthria). อาการอื่นๆ ของสมองส่วนหลัง ดังเช่นปัญหาในการกลอกตา อาการสั่น (kinetic tremor) สามารถเกิดขึ้นได้เช่นกัน แต่พบได้น้อยกว่าอาการของสมองส่วนหลัง มักเป็นอาการร่วมกับอาการพาร์กินโซนิซึม แต่มักจะสังเกตได้เด่นชัดในผู้ป่วยกลุ่ม MSA-C.
ผู้ป่วย MSA สามารถมีอาการร่วมอื่นๆ นอกเหนือจากอาการทางระบบประสาทอัตโนมัติพาร์กินโซนิซึม และอาการของสมองส่วนหลัง. อาการทาง pyramidal signs ดังเช่น brisk tendonreflexes, spasticity, extensor plantar responses และ pseudobulbar palsy มักพบได้บ่อย และสามารถตรวจพบได้ในช่วงแรกของผู้ป่วย MSA. อาการ dementia ถึงแม้ว่าจะพบน้อย และไม่เด่นชัด ถ้าเกิดขึ้นจะเป็นลักษณะอาการของ frontal lobe โดยส่วนใหญ่การวินิจฉัยโรค MSA จะอาศัยลักษณะอาการดังที่กล่าวไว้ข้างต้น ร่วมกับ diagnostic criteria ที่ได้กำหนดขึ้น.13 การตรวจทางห้องปฏิบัติการ โดยเฉพาะการตรวจ MRI จะช่วยบ่งบอกถึงรอยโรค และสนับสนุนลักษณะอาการที่เด่นในผู้ป่วยรายนั้นว่ามีพยาธิสภาพชัดเจนในสมองส่วนใด ดังเช่น MRI ในผู้ป่วย MSA-C มักปรากฏลักษณะของ brainstem and cerebellar atrophy. ลักษณะผิดปกติทาง MRI ที่พบได้มากกว่าร้อยละ 50 ของผู้ป่วย MSA โดยทั่วไป โดยเฉพาะในกลุ่ม MSA-P คือ hypointense signals on T2-weighted images เนื่องจากการสะสมของธาตุเหล็ก ร่วมกับ slit-like hyperintense signals on T2-weightd images ในส่วนของ putamen ส่วนนอก ลักษณะความผิดปกติของสมองส่วน Pons ซึ่งเกิดเนื่องจากการเสื่อมของ transversed pontine fibers และ midline raphe โดยที่ tegmentum ปกติ ทำให้ภาพของ MRI ในสมองส่วนนั้นมีลักษณะคล้าย Hot-cross bun (ภาพที่ 8)
นอกเหนือจากการตรวจทางรังสีวิทยาที่พบความผิดปกติดังกล่าว การตรวจทางกล้ามเนื้อ และเส้นประสาท (EMG & NCS) อาจพบความผิดปกติของเส้นประสาทส่วนปลายโดยที่ผู้ป่วยไม่มี
อาการแสดงออก (subclinical peripheral neuropathy) หรือการเสื่อมของหูรูด กระเพาะปัสสาวะ และทวารหนัก (denervation of external urethral and anal sphincter). การตรวจ tilt table อาจพบความผิดปกติในเรื่องของความดันโลหิต (postural hypotension) เป็นต้น.
ลักษณะทางพยาธิวิทยาในผู้ป่วย MSA จะพบความผิดปกติกระจายในสมองหลายๆ ส่วนที่นอกเหนือจากส่วนของ substantia nigra ดังเช่น striatum, locus coeruleus, Edinger-Westphal nucleus, dorsal motor nucleus of vagus nerve, middle cerebellar peduncles, inferior olive, intermediolateral cell columns และ Onuf's nucleus ซึ่งความผิดปกติ ประกอบด้วย Glial cytoplasmic inclusions (GCIs) กระจายอยู่รอบ nucleus ของ oligodendroglia ในสมองหลายๆ ส่วน ที่ได้กล่าวไปข้างต้น. cytoplasmic inclusions และ neuronal nuclear inclusions อื่นๆ ก็สามารถพบได้เช่นกัน โดยเฉพาะเมื่อทำการย้อมพิเศษด้วย alpha-synuclein antibody.
3. Corticobasal degeneration (CBD)
ดังชื่อที่ปรากฏ อาการของ CBD รวมถึงอาการของ cortical dysfunction ซึ่งโดยส่วนใหญ่จะเป็นอาการของ cortical sensory loss หรือ apraxia ของร่างกายด้านใดด้านหนึ่ง ร่วมกับอาการพาร์กินโซนิซึมในข้างเดียวกัน ซึ่งลักษณะอาการดังกล่าว ร่วมกับการไม่ตอบสนองต่อยาลีโวโดปา14 จะเป็นลักษณะที่ช่วยแยก CBD ออกจากโรคพาร์กินสัน. อาการแรกเริ่มในผู้ป่วยกลุ่มนี้มักเป็นปัญหาในการเคลื่อนไหวช้าของแขนขาด้านใดด้านหนึ่งร่วมกับปัญหาในการรับความรู้สึก หรือการสั่งงานของแขนขาด้านนั้น ซึ่งอาการทั้งหมดที่กล่าวไป มักจะเกิดภายใน 3 ปีแรก. นอกจากนี้ผู้ป่วยอาจมีอาการอื่นๆ ดังเช่น อาการปวดในแขนขาด้านนั้น ซึ่งอาจเกิดมากกว่าปกติ เมื่อเทียบกับสิ่งที่กระตุ้น (allodynia) อาการเกร็งหรือดีสโตเนีย และอาการกระตุก (myoclonic jerks) ของแขนขาด้านนั้น. เมื่อเวลาผ่านไปอาการดังกล่าวสามารถเกิดได้ในด้านตรงข้าม แต่อย่างไรก็ตาม ความรุนแรงมักจะน้อยกว่าด้านแรกที่เริ่มมีอาการ และปัญหาในเรื่องของการเดินซึ่งมักจะเกิดเฉพาะในระยะหลัง ซึ่งเป็นอาการที่ช่วยแยก CBD ออกจาก PSP หรือ MSA ซึ่งมักจะมีปัญหาในการเดิน และการทรงตัวในระยะแรก.
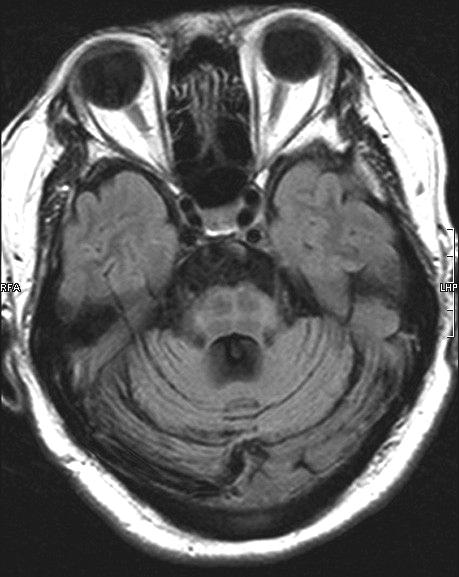
ภาพที่ 8. Hot-cross bun sign.
จากการวิจัยที่รวมผู้ป่วย CBD เพื่อวิเคราะห์ถึงลักษณะอาการที่จะช่วยในการวินิจฉัยโรค CBD ปรากฏว่าอาการเริ่มแรกได้แก่ อาการดีสโตเนีย ด้านใดด้านหนึ่งร่วมกับอาการพาร์กินโซนิซึมในด้านเดียวกัน ideomotor apraxia และการเดินที่เป็นปกติ จะเป็นอาการที่ช่วยบ่งชี้โรค CBD. อาการสั่นในโรค CBD เกิดน้อย โดยส่วนใหญ่เป็น action หรือ postural tremor ที่เร็ว ซึ่งอาจเกิดร่วมกับ myoclonus ในด้านเดียวกัน ต่างจากอาการสั่นในโรค พาร์กินสันที่โดยส่วนใหญ่เป็นอาการสั่นในขณะมืออยู่เฉย (asymmetric rest tremor).
Alien limb phenomenon หมายถึง การที่ผู้ป่วยไม่สามารถบอกว่าแขนหรือขาส่วนนั้น เป็นส่วนของร่างกายตนเอง พบได้ประมาณร้อยละ 50 ของผู้ป่วย CBD แต่มักเป็นอาการที่ตามมาภายหลังร่วมกับอาการปวดของแขนขาด้านนั้น ความผิดปกติของการกลอกตาในผู้ป่วย CBD จะเป็นในเรื่องของ increased latency of horizontal saccades ซึ่งต่างจากผู้ป่วย PSP ที่จะมี saccades ที่ช้าลง นอกจากนี้ pursuit eye movement จะช้าลงร่วมกับ saacadic breakdown โดยที่การมองไปทางด้านข้าง หรือขึ้นลง ยังสามารถทำได้เต็ม 180 องศา.
ผู้ป่วย CBD มักมีอาการเด่นชัดของ cortical dysfunction ซึ่งโดยส่วนใหญ่เป็นอาการ apraxia, cortical sensory loss และ dementia. ในส่วนของ apraxia โดยส่วนใหญ่จะเป็นในลักษณะของ ideomotor apraxia ซึ่งหมายถึงการที่ผู้ป่วยไม่สามารถใช้แขนขาด้านที่ผิดปกติในการเลียนแบบท่าทาง ซึ่งปัญหาไม่ได้เกิดจากการอ่อนแรงหรือการไม่ได้รับความรู้สึกของแขนขาด้านนั้น. Apraxia แบบ อื่นๆ ได้แก่ ideational และ limb-kinetic apraxia พบได้น้อยกว่า.
การวินิจฉัยโรค CBD โดยส่วนใหญ่อาศัยลักษณะอาการตามที่ได้กล่าวไว้ข้างต้น ผู้ป่วยโดยส่วนใหญ่จะมีอาการแสดงออกที่ชัดเจนเมื่อระยะเวลาของโรคนานขึ้น ทำให้การวินิจฉัยมักไม่เป็นปัญหาในผู้ป่วยช่วงระยะหลังๆ การตรวจทางห้องปฏิบัติการโดยส่วนใหญ่จะปกติ. การตรวจทางรังสีวิทยา โดยเฉพาะ MRI จะเป็นปกติในช่วงแรกของโรค เมื่อระยะเวลาของโรคนานขึ้น ลักษณะฝ่อของสมองในส่วน posterior frontal และ parietal lobes มักจะชัดเจนขึ้น โดยเฉพาะในด้านตรงข้ามกับแขนขาส่วนที่มีอาการร่วมกับ abnormal signal attenuation ในส่วนของ subcortical white matter ที่ติดกับสมองส่วนที่ฝ่อลง.
เช่นเดียวกับความผิดปกติที่พบใน MRI ลักษณะทางพยาธิวิทยาในโรค CBD แสดงถึงเนื้อสมองที่บาง และฝ่อลงในส่วนของ posterior frontal และ parietal lobes ในด้านตรงข้ามกับแขนขาที่มีอาการการฝ่อจะพบมากที่สุดในส่วนของ medial perirolandic superior frontal gyrus, parasagittal pre-and postcentral gyri และ superior parietal lobule. ความผิดปกติทางจุลพยาธิวิทยาจะเป็นในลักษณะของ vacuolar change และ spongiosis ในส่วนของผิวสมอง (upper cortical layers) ร่วมกับ ballooned neuron ซึ่งเกิดเนื่องจาก axonal damage และการย้อม silver stain จะพบ Neurofibrillary tangles (NFTs) ในส่วนของสมองที่มีความผิดปกติดังกล่าว รวมถึงในสมองส่วน substantia nigra โดยไม่พบ neuritic plaques (ซึ่งเป็นลักษณะทางพยาธิวิทยาในโรคอัลไซเมอร์).
4. Diffuse Lewy body disease
โรค Dementia with Lewy bodies หรือที่แพทย์บางท่านใช้คำว่า Diffuse Lewy body disease (DLB) มีอาการที่แตกต่างจากผู้ป่วยโรคพาร์กินสันในลักษณะของอาการเด่นทางด้าน cognitive dysfunction ร่วมกับอาการพาร์กินโซนิซึม. อาการ cognitive dysfunction จะเป็นในลักษณะของการเห็นภาพหลอน อาการสับสน และอาการสับสนทางจิตเวช (psychosis). นอกจากนี้ อาการดังกล่าวอาจเกิดเป็นช่วงๆ สลับกับเวลาที่ผู้ป่วยรู้สึกตัวดี ร่วมกับการหกล้มที่ค่อนข้างบ่อย หรืออาการหมดสติไปเป็นช่วงๆ โดยที่ไม่ทราบสาเหตุ. อาการเหล่านี้สามารถเกิดขึ้นได้เอง หรือมีอาการมากขึ้นเมื่อผู้ป่วย ได้รับยา dopaminergic drugs หรือยาในกลุ่ม neuroleptics (dopaminergic and neuroleptics hypersensitivity). อาการพาร์กินโซนิซึม โดยส่วนมากจะเป็นในลักษณะของอาการแข็งเกร็ง เคลื่อน ไหวช้าที่เป็นทั้ง 2 ด้านพร้อมๆ กัน.
การแยกโรคระหว่าง DLB และโรคพาร์กินสันอาจเป็นไปได้ยาก ผู้เชี่ยวชาญบางท่านยังคงเชื่อว่าโรค DLB อาจจะเป็นส่วนหนึ่งของโรคพาร์กินสัน ไม่ใช่โรคต่างหาก. แต่โดยทั่วไป อาการทาง cognitive dysfunction ในผู้ป่วยโรคพาร์กินสันจะเกิดช้า ส่วนใหญ่มากกว่า 5-10 ปีแล้วขึ้นไป ถ้าอาการใน เรื่อง cognitive dysfunction เด่น เกิดขึ้นภายใน 1 ปีแรกของอาการพาร์กินโซนิซึม โอกาสที่ผู้ป่วย จะเป็นโรค DLB ก็ค่อนข้างสูง. ในทางพยาธิวิทยาของโรค DLB จะพบ Lewy bodies ในส่วนของ brainstem, neocortex, cingulated cortex และ transentorhinal cortex ร่วมกับ Lewy-body neuritis, plaques, neurofibrillary tangles ต่างจากพยาธิวิทยาในโรคพาร์กินสัน ซึ่ง Lewy bodies จะพบมากในส่วนของ substantia nigra pars comtacta, locus coeruleus, dorsal motor nucleus of the vagus และ basal nucleus of Meynert. การแยกทางพยาธิวิทยาระหว่าง DLB และโรคอัลไซเมอร์อาจแยกได้ลำบาก.
การรักษาอาการพาร์กินโซนิซึม
เนื่องจากพาร์กินโซนิซึมเป็นอาการเกิดขึ้นได้เนื่องจากสาเหตุต่างๆ ตามที่ได้กล่าวไว้ข้างต้น ดังนั้นการรักษาที่สำคัญ คือการรักษาตามสาเหตุนั้นๆ ดังเช่น ถ้าอาการพาร์กินโซนิซึมเกิดเนื่องจากยา metoclopramide การรักษาที่สำคัญ คือการหยุดการใช้ยานั้น ร่วมกับการสังเกตอาการอย่างต่อเนื่อง.
โดยทั่วไป การตอบสนองต่อยาลีโวโดปา เป็นวิธีหนึ่งที่แพทย์ใช้ในการวินิจฉัยแยกโรคผู้ป่วยพาร์กินโซนิซึม โดยที่ผู้ป่วยโรคพาร์กินสันจะมีการตอบสนองที่ดีต่างจากกลุ่มโรคพาร์กินโซนิซึมอื่นๆ โดยเฉพาะผู้ป่วยโรคพาร์กินสันเทียมที่มักมีการตอบสนองที่ไม่ดี. ทั้งนี้สาเหตุของความแตกต่างส่วนหนึ่งเกิดจากลักษณะทางพยาธิวิทยาของแต่ละโรคที่มีความแตกต่างกันไป. ในโรคพาร์กินสัน พยาธิสภาพโดยส่วนใหญ่จะอยู่ที่ presynaptic dopamine ซึ่งเกิดเนื่องจากการเสื่อมของเซลล์ในส่วนของ substantia nigra pars compacta เป็นหลักใหญ่ ทำให้เกิดภาวะของการขาดสารโดปามีน โดยที่ส่วนของ postsynaptic dopamine (dopaminergic receptors) ค่อนข้างปกติ. ดังนั้น ผู้ป่วยพาร์กินสันเมื่อได้รับยาลีโวโดปา ซึ่งเปลี่ยนเป็นโดปามีนในสมอง ผู้ป่วยจึงมีอาการดีขึ้น อาการพาร์กินโซนิซึมที่ลดลง. ในทางกลับกัน โรคพาร์กินสันเทียมอื่นๆ โดยส่วนใหญ่มีพยาธิสภาพที่ค่อนข้างกระจัดกระจาย ทั้ง Pre-& postsynaptic dopamine dopamine receptors มีการเสื่อมเช่นเดียวกัน ดังนั้นถึงแม้ผู้ป่วยโรคพาร์กินสันเทียมได้รับลีโวโดปา ซึ่งสามารถเปลี่ยนเป็นโดปามีน ในผู้ป่วยนั้น โดปามีนก็ไม่สามารถไปจับกับ dopamine receptors ได้ ทำให้ผู้ป่วยโรคพาร์กินสันเทียม โดยส่วนใหญ่ไม่มีอาการดีขึ้นอย่างชัดเจน หรือตอบสนองเป็นเพียงบางส่วนเท่านั้น.
นอกเหนือจากการตอบสนองต่อยาลีโวโดปาที่จะใช้ช่วยแยกโรคพาร์กินสันออกจากโรคพาร์กินสันเทียม. การเกิดการตอบสนองต่อยาที่ไม่สม่ำเสมอ หรือ motor fluctuations เป็นอาการที่ค่อนข้างเฉพาะในโรคพาร์กินสัน เมื่อได้รับการรักษาด้วยยาในกลุ่มโดปามีนอย่างไม่สม่ำเสมอ เนื่องจากการกระตุ้นเป็นช่วงๆ จากการปลดปล่อยโดปามีนเป็นช่วงๆ (pulsatile stimulation) เนื่องจากการเสื่อมของส่วน presynaptic dopamine ในโรคพาร์กินสัน. ปัญหาดังกล่าวมักไม่เกิดในกลุ่มโรคพาร์กินโซนิซึมอื่นๆ หรือถ้าเกิดก็จะน้อยและไม่รุนแรง ซึ่งสามารถอธิบายได้คร่าวๆ เนื่องจากพยาธิสภาพในส่วนของ postsynaptic dopamine หรือ dopamine receptors ในกลุ่มโรคพาร์กินโซนิซึมอื่นๆ ซึ่งสาเหตุโดยละเอียดของ motor fluctuations นั้นยังมีมากกว่าโดปามีนซึ่งไม่ได้กล่าวรายละเอียดไว้ในบทความนี้.
รุ่งโรจน์ พิทยศิริ พ.บ., M.R.C.P.(UK.)
American Board of Psychiatry & Neurology
ผู้ช่วยศาสตราจารย์, ศูนย์รักษาโรคพาร์กินสันและกลุ่มโรคความเคลื่อนไหวผิดปกติ,
สาขาวิชาประสาทวิทยา, คณะแพทยศาสตร์, จุฬาลงกรณ์มหาวิทยาลัย