-
ขนาดตัวอักษร
 |
|
 |
|
 |
|

ABC Transporters and Drug Therapy
ในปัจจุบันการศึกษาประสิทธิผลของยาในการรักษา สามารถสรุปความเกี่ยวเนื่องกันได้เป็นสองกระบวนการใหญ่ๆ คือ เภสัชจลนศาสตร์ (pharmacokinetics) และเภสัชพลศาสตร์ (pharmacodynamics) ในการบริหารยาโดยวิธีรับประทานนั้น ฤทธิ์ทางเภสัชวิทยาส่วนใหญ่มักขึ้นกับปริมาณที่ยาสามารถถูกดูดซึมผ่านลำไส้อย่างเพียงพอเข้าสู่ร่างกาย และมีการกระจายตัวไปยังตำแหน่งของการออกฤทธิ์ ก่อนที่จะถูกกำจัดออกจากร่างกายโดยกระบวนการ metabolism และการขับยา (excretion) ออกจากร่างกายผ่านอวัยวะต่างๆเช่น ตับ และไต.
การศึกษาเกี่ยวกับ drug disposition ส่วนใหญ่มักจะมุ่งศึกษาถึงบทบาทของ drug metabolizing enzymes เป็นหลัก ในการเกิดปฏิกิริยา metabolism โดยทั่วไปมักจะแบ่งง่ายๆ ออกเป็น 2 phase คือ phase I reaction เช่น oxidation, reduction และ hydrolysis ซึ่งส่วนใหญ่มักจะผ่านเอนไซม์ Cytochrome P450 (CYP) family และใน phase II metabolism จะเกิดปฏิกิริยาผ่าน endogenous compounds เช่น glucuronic acid, glutathione หรือ sulfate ในการ conjugate ยาหรือ metabolite จาก phase I เพื่อทำให้กลายเป็นสารที่มีขั้ว (polar) มากขึ้นซึ่งจะถูกกำจัดออกจากร่างกายได้เร็วขึ้น.
คุณสมบัติทางเคมีและกายภาพของยา เช่น ค่า pKa, ขนาดอนุภาค และ lipophilicity เป็นปัจจัยที่มีผลต่อความสามารถในการถูกดูดซึมเข้าสู่ร่างกาย และ target tissue compartment ข้อมูลในปัจจุบันพบว่ากระบวนการขนส่งยาเข้าสู่ร่างกายโดย carrier mediated processes หรือ transporters เป็นอีกปัจจัยที่มีความสำคัญ โดยขึ้นกับระดับของการแสดงออก (expression) ของ transporters ในอวัยวะต่างๆ เช่น ลำไส้ ตับ และไต เป็นต้น และmembrane transporters เป็นโปรตีนที่มีบทบาทสำคัญในการควบคุม physiologic solute และสมดุลของเหลวในเซลล์ โดยจากผลการศึกษาจีโนมในมนุษย์ พบว่ามีประมาณ 500-1,200 gene encode transport proteins.
การทำงานของ protein transporter แต่ละชนิดก็แตกต่างกันไป และในบางชนิดก็ทำหน้าที่ได้หลายอย่างรวมทั้งมีบทบาทสำคัญทางสรีรวิทยาในการขนส่ง endogenous substances ต่างๆ เช่น น้ำตาล, ไขมัน, amino acid, bile acid, steroids และฮอร์โมนต่างๆ ในร่างกาย.
ในทางเภสัชวิทยา เราสามารถแบ่ง drug transporters ได้เป็นสองกลุ่มใหญ่ คือ กลุ่มที่เป็น uptake transporters และ กลุ่มที่เป็น efflux transporters.1
Uptake transporters ทำหน้าที่เพิ่มความสามารถในการนำส่งยาเข้าสู่เซลล์ ซึ่งมี transporters หลายกลุ่ม ได้แก่ organic anion transporting polypeptide (OATP, SLCO) family, organic anion transporter (OAT, SLC22A) และ organic cation transporter (OCT, SLC22A) family, organic cation/carnitine transporter (OCTN, SLC22A) family และpeptide transporter (PEPT, SLC15A) family.
กลุ่มที่สองคือ Efflux transporters ทำหน้าที่ส่งยาออกจากเซลล์ไปสู่นอกเซลล์โดยต้านกับ concentration gradient สมาชิกส่วนใหญ่ของ transporters กลุ่มนี้อยู่ใน adenosine triphosphate (ATP)-binding cassette (ABC) superfamily ของ transmembrane proteins ซึ่งอาศัยพลังงานจาก ATP hydrolysis ในการนำส่งยาผ่าน biologic membrane ตัวอย่าง protein transporters ในกลุ่มนี้ได้แก่ P-glycoprotein (ABCB) family เช่น multidrug resistance protein 1 (MDR1), กลุ่มของ bile salt export pump (BSEP), multidrug resistance-associated (MRP, ABCC) protein family, และ breast cancer resistance protein (BCRP, ABCG).
นอกจากนี้ ยังมี protein transporters กลุ่มอื่นๆ อีก บางชนิดสามารถเป็นทั้ง influx และ efflux transporters เช่น ในกลุ่ม OATPs ซึ่งจะทำหน้าที่เป็น uptake transporters ได้โดยผ่าน sodium-independent mechanism และเป็น efflux tran-sporters โดยทำหน้าที่เป็น sodium-dependent uptake transporter เช่น sodium-taurocholateco-transporting polypeptide (NTCP) เป็นต้น.2
เนื่องจากบทบาทของ protein transporters ในกลุ่ม ABC transporters ที่เกี่ยวข้องกับการขนส่งยาในร่างกายเป็นที่กล่าวถึงกันมากขึ้นและมีการศึกษาหน้าที่การทำงาน การควบคุมการทำงาน และการแสดงออกของ protein transporters ในอวัยวะต่างๆ เพื่อประยุกต์ใช้ทางคลินิกกันอย่างกว้างขวาง. บทความนี้จะนำเสนอบทสรุปเกี่ยวกับบทบาทของ ABC transporters ต่อเภสัชจลนศาสตร์ของยาในภาพรวมที่เกี่ยวข้องกับ protein transporters ใน การรักษามะเร็งที่ดื้อต่อยาเคมีบำบัด จากข้อมูลที่ได้จากงานวิจัยต่างๆ จากการวิจัยแบบ in vitro และการศึกษาวิจัยในมนุษย์ เพื่อนำไปสู่แนวคิดแนวทางเพื่อประยุกต์ใช้ในงานวิจัยทางคลินิกต่อไป.
ABC Transporters
การศึกษาในมนุษย์ได้มีการค้นพบหน่วยพันธุกรรมของ human ABC gene ถึง 48 ชนิด ซึ่งมีความคล้ายคลึงกันใน sequence และ structural homology ส่วนชนิดที่สำคัญและมีการศึกษาถึงผลต่อ pharmacokinetics ของยาได้แก่ P-glycoprotein (MDR1, ABCB), multidrug resistance-asso- ciated (MRP, ABCC) protein (ในปัจจุบันพบว่ามี MRP1 ไปจนถึง MRP8) และ breast cancer resistance protein (BCRP, ABCG).3
การกระจายตัวและการแสดงออก (expression) ของ protein transporters เหล่านี้แตกต่างกันไปตามอวัยวะหรือเนื้อเยื่อ (ตารางที่ 1) นอกจากนี้ protein transporters เหล่านี้ยังมีความจำเพาะและมีหน้าที่ในการส่งผ่าน endogenous และ exogenous substrates ที่แตกต่างกันอีกด้วย (ตารางที่ 2). 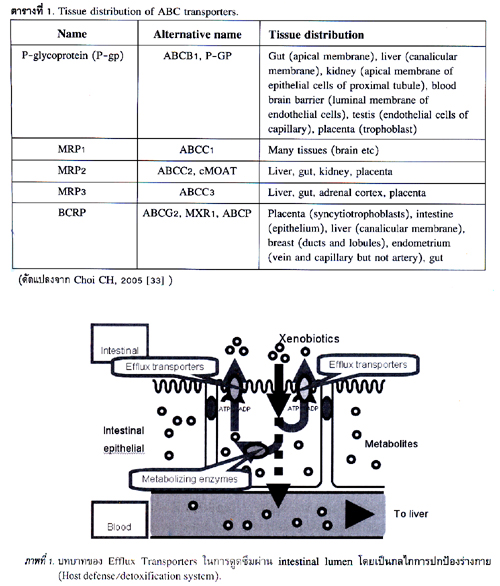
ธรรมชาติของ ABC transporters มีความคล้ายคลึงกันกับกรณีของ cytochrome P450 isoenzymes หลายๆชนิด พบว่า ABC transporters โดยเฉพาะอย่างยิ่ง P-glycoprotein มี genetic polymorphisms คือ มีรูปแบบของยีนในการแสดงออกทางพันธุกรรมที่ตำแหน่งเดียวกันแตกต่างกันไปในกลุ่มประชากร ซึ่งอาจส่งผลต่อการตอบสนองต่อยา และ pharmacokinetics ของยา ทำให้มีโอกาสเกิดพิษจาก xenobiotics (สารแปลกปลอมต่างๆ จากภายนอก ที่ได้รับเข้าสู่ร่างกาย เช่น ยาสารพิษสารเคมีต่างๆ) แตกต่างกันไปในแต่ละบุคคลได้.4 อย่างไรก็ตามรายละเอียดข้อมูลการศึกษา genetic polymorphisms ในมนุษย์ยังมีไม่มากนัก และยังไม่สามารถสรุปผลทางคลินิกได้ มีการศึกษาหนึ่งที่แสดงให้เห็นถึงความแตกต่างอย่างชัดเจนของ genotype และ allele frequency ของ MDR1 gene (ยีนที่กำหนดการแสดงออกของ P-glycoprotein ซึ่งมี genotype แบบ C/C, C/T, และ TT โดย C คือ wild-type allele และ T คือ mutant allele). การศึกษาดังกล่าวพบว่าในกลุ่มชาวแอฟริกัน ประกอบด้วย Ghanaian, Kenyan, African-American และ Sudanese มี allele frequency ของ C/C allele (คือมี genotype เป็นแบบ homozygous wild-type) อยู่ระหว่างร้อยละ 73-84 และเมื่อเปรียบเทียบกับประชากรกลุ่ม Caucasian/Asian ซึ่งประกอบด้วย British Caucasian, Portuguese, Southwest Asian, Chinese, Filipino and Saudi ซึ่งพบว่ามี allele frequency ของ C/C allele อยู่เพียงร้อยละ 34-55 เท่านั้น. แม้ว่าในการศึกษานี้ไม่ได้มีการวิเคราะห์ระดับ expression ของ P-glycoprotein โดยตรงก็ตาม แต่ผลการศึกษาดังกล่าว ก็พอจะอนุมานถึง expression ของ P-glycoprotein ในกลุ่มประชากรแอฟริกัน ว่าน่าจะมีแนวโน้มสูงในกลุ่มประชากร Caucasian/Asian5 หากข้อสันนิษฐานดังกล่าวเป็นจริง เรื่องนี้อาจโยงไปถึงการอธิบายว่ายา ivermectin (antihelmintic agent) ที่เป็น substrate ของ P-glycoprotein ซึ่งยานี้มีความเป็นพิษต่อระบบประสาทในสัตว์ทดลองที่มี P-glycoprotein expression ต่ำ แต่กลับสามารถใช้ได้อย่างปลอดภัยโดยไม่พบการเกิดพิษดังกล่าวในการรักษาโรค river blindness (onchocerciasis) ในแอฟริกาซึ่งในปัจจุบันมีการใช้ยานี้ในผู้ป่วยชาวแอฟริกันราวกว่า 20 ล้านคนแล้ว แต่ก็ยังไม่มีรายงานเกิดพิษต่อระบบประสาทในกลุ่มประชากรดังกล่าว และนอกจากนี้ การมี expression ของ P-glycoprotein สูงในชาวแอฟริกัน ก็อาจจะสามารถโยงไปเกี่ยวข้องกับการที่พบว่ามีอุบัติการณ์ของมะเร็งที่ดื้อต่อการรักษาด้วยยาเคมีบำบัดสูงในผู้ป่วยกลุ่มดังกล่าวด้วย.6
Effects of ABC transporters on Pharmacokinetics
บทบาทสำคัญของ ABC transporters ใน การเป็น efflux transporter ที่เกี่ยวข้องกับ pharmacokinetics ทำให้เกิดความสนใจในการทำวิจัยเกี่ยว กับเรื่องนี้กันมากขึ้น แม้ว่าความรู้ในปัจจุบันเกี่ยวกับหน้าที่การทำงานที่แท้จริงของ ABC transporters ในร่างกายมนุษย์จะยังไม่ทราบทั้งหมดก็ตาม.
ในมนุษย์เราจะพบ P-glycoprotein ที่บริเวณ apical surface ของ columnar epithelial cells ของทั้งลำไส้เล็กและลำไส้ใหญ่, biliary canalicular membrane ของ hepatocyte, apical surface ของ epithelial cells ของ proximal tubules ในไต, apical surface ของ epithelial cells ในรก, และที่ apical surface ของ endothelial cells ใน blood capillaries ของสมอง.7,8
จากตำแหน่งที่ ABC transporters ปรากฏบนอวัยวะที่สำคัญเหล่านั้น ทำให้มีบทบาทในการจำกัดปริมาณยา หรือ xenobiotics จาก intestinal lumen ไปสู่ enterocytes รวมทั้งการผ่านของยาจากระบบ เลือดเข้าไปสู่สมอง และทางรก (placenta) นอกจากนี้ ยังไปมีบทบาทในการเพิ่มหรือเร่งการกำจัดยาออกจาก hepatocytes, renal tubule, intestinal epithelial cells ไปสู่ luminal spaces และขับออกไปจากร่างกาย.
จากกระบวนการทั้งหมด ABC transporters จึงน่าจะมีหน้าที่หลักสำคัญประการหนึ่ง ในการปกป้องเซลล์ในร่างกายจากอันตรายที่จะได้รับจากยาต่างๆ, xenobiotics หรือสารพิษต่างๆ ในสิ่งแวดล้อม และในบรรดา ABC transporters ทั้งหมด พบว่า P-glycoprotein เป็น efflux transporters ที่มีบทบาทมากที่สุดในการขับยากลับออกมาสู่ intestinal lumen ทำให้มีผลกระทบอย่างมากต่อ bioavailability ของยาที่เป็น substrates. จากการศึกษา oral bioavailability ของ paclitaxel, digoxin และ HIV protease inhibitors ซึ่งเพิ่มขึ้นสูงกว่าใน mdr1a knockout mice เมื่อเปรียบเทียบกับ wild-type mice ซึ่งพบว่า bioavailability ของยาต่ำกว่ากลุ่ม wild-type อย่าง มีนัยสำคัญ9-11 และการศึกษาในมนุษย์สามารถยืนยันผลดังกล่าวจากการศึกษาผลของระดับ MDR1 expression ในลำไส้ต่อระดับยา digoxin และ cyclo-sporine ในเลือด.12,13 อย่างไรก็ตาม ในทำนองเดียวกันกับ drug metabolism enzymes มีการศึกษาพบว่า efflux function ของ P-glycoprotein ในทางเดินอาหารก็อาจมีการอิ่มตัว (saturated) ได้. ในกรณีที่ความเข้มข้นของยาใน intestinal lumen สูงเกินกว่าค่า Km (Michaelis-Menten Constant) จากการกินยาในขนาดสูง ดังตัวอย่างที่ทำการศึกษาในเนื้อเยื่อลำไส้มนุษย์ (colon) ซึ่งพบว่า digoxin มีค่า Km เท่ากับ 59 mmol/Lเป็นต้น.14
Efflux transporters ที่อยู่บริเวณ canalicular (apical) membrane ของ hepatocyte เช่น MDR1, MRP2, and BCRP จะมีบทบาทในช่วงสุดท้ายของการส่งผ่านยาจากตับออกสู่น้ำดี และนอกเหนือไปจาก P-glycoprotein ที่กล่าวมาแล้ว canalicular multi-specific organic anion transporter หรือ MRP2 ก็มีบทบาทสำคัญในการขับ endogenous organic anions เช่น bilirubin glucuronides รวมทั้งยา methotrexate, irinotecan (CPT-11) และ pravastatin ออกทางน้ำดีอีกด้วย.15-17
บทบาทของ protein transporters ที่ไตนั้นมีการทำงานร่วมกันของ uptake และ efflux transporters ที่บริเวณ basolateral และ apical membrane ของ proximal tubular cells โดยพบว่าบทบาทส่วนใหญ่ในการ uptake ของ organic anion เกิดจาก OAT family ซึ่งมีผลต่อยาที่เป็น substrate ของ transporters กลุ่มนี้ เช่น ยาในกลุ่ม b-lactam antibiotics, diuretics, nonsteroidal anti-in- flammatory drugs (NSAIDs), nucleoside antiviral drugs และยาในกลุ่ม anticancer agents เป็นต้น.18 ส่วน renal efflux transporters ยังไม่ได้มีการศึกษากันมากนัก แต่ก็พบว่า OATP families และ MRP ที่อยู่บริเวณ apical membrane ของ proximal tubular cell เป็นกลุ่มหลักที่มีหน้าที่เกี่ยวข้องกับการกำจัดยาที่เป็น substrate ทางปัสสาวะ.19
จากผลของ protein transporters ที่มีต่อ pharmacokinetics ของยา ดังที่กล่าวมาแล้ว จะเห็นว่า หากมีการได้รับยาหรือสารที่มีฤทธิ์เปลี่ยนแปลงการทำงานของ protein transporters ร่วมกับยาที่เป็น substrates ของ transporters นั้นๆ แล้วย่อมมีผลกระทบในรูปแบบของ drug interaction ซึ่งอาจเกิดอาการพิษ หรืออาการไม่พึงประสงค์จากยาที่เป็น substrate ของ protein transporters ชนิดนั้น เช่นกรณีที่ได้รับยาที่เป็น P-glycoprotein inhibitor มีผลให้ระดับยาในเลือดของยาที่เป็น substrate ในร่างกายสูงขึ้นจนเกินระดับการรักษา (therapeutic range) จนเกิดพิษจากยาได้. อย่างไรก็ตาม ปฏิกิริยาต่อกันดังกล่าว ก็อาจมีประโยชน์ในการพัฒนาการขนส่งยาไปถึงยังอวัยวะเป้าหมายได้ดีขึ้น ดังเช่น การพัฒนา P-glycoprotein inhibitor มาทดลองใช้เสริมในการรักษามะเร็งที่ดื้อต่อยาเคมีบำบัด เป็นต้น. ตัวอย่างสารที่เป็น inhibitors ของ protein transporters ชนิดต่างๆ แสดงในตารางที่ 2.
ในอีกกรณี xenobiotics หรือยาที่ได้รับร่วมกัน อาจส่งผลกระทบต่อประสิทธิภาพในการออกฤทธิ์ของยาที่อวัยวะเป้าหมายได้ เช่น การได้รับยาที่เป็น P-glycoprotein inducer ทำให้ expression ของ P-glycoprotein มีมากขึ้นอาจทำให้ยาถูกผลักออกจากเซลล์กลับสู่ทางเดินอาหารมากขึ้นและมี oral bioavailability ลดลง ตัวอย่างของ P-glycoprotein inducer ได้แก่ dexamethasone, cyclosporin, rifampicin, phenobarbital, clotrimazole, reserpine รวมทั้งสารสกัดที่ได้จากสมุนไพร เช่น St John's Wort เป็นต้น. อย่างไรก็ตามการนำผลการศึกษาเหล่านี้ไปใช้ทางคลินิกอาจต้องพิจารณาเพิ่มเติมในเรื่องขนาดยาและการตอบสนองของ species ที่แตกต่างกันร่วมด้วย ดังเช่นในการศึกษาเกี่ยวกับผลของ St John's Wort เนื่องจากพบว่าสารสกัดมีผลต่อการเพิ่ม expression ของ P-glycoprotein ในหนูทดลอง มากกว่าในมนุษย์อาสาสมัครสุขภาพดี แต่เมื่อพิจารณา ขนาดยาที่หนูทดลองได้รับ พบว่าขนาดยาสูงมากกว่าที่อาสาสมัครในการศึกษาได้รับประมาณ 50 เท่า เมื่อคิดเทียบขนาดยาต่อน้ำหนักตัวเป็นกิโลกรัม.20
นอกจากนี้ เป็นที่น่าสังเกตว่าสารที่มีผลเป็น P-glycoprotein inducer เหล่านี้มักมีการคาบเกี่ยวกันกับการมีคุณสมบัติเป็น potent CYP3A4 inducer ร่วมด้วย และยังพบว่าการเหนี่ยวนำให้เพิ่ม expression ของ P-glycoprotein นั้น การตอบสนองจะมีความจำเพาะ และแตกต่างกันในแต่ละอวัยวะ ดังตัวอย่างการศึกษาในหนูทดลองที่ได้รับ cyclosporine ในขนาด 10 มก./กก./วัน เป็นเวลา 15 วัน พบว่าสามารถเพิ่ม expression ของ P-glycoprotein ในไต ตับ ลำไส้ กระเพาะอาหาร และปอด ซึ่งมีค่าเพิ่มขึ้นประมาณร้อยละ 70-250 โดยมีการตอบสนองสูงสุดหลังจากการได้รับยาอย่างต่อเนื่องกันในวันที่ 10 ส่วนที่หัวใจ อัณฑะ และม้าม พบว่าเพิ่มขึ้นเพียงร้อยละ 50-80 และตอบสนองสูงสุดใน 5 วัน แต่กลับไม่พบความเปลี่ยนแปลงของ P-glycoprotein expression ที่ blood brain barrier ในสมอง ซึ่งอาจมีสาเหตุมากจากการที่สมองมี expression ของ P-glycoprotein ที่สูงอยู่ก่อน จึงสามารถ efflux cyclosporine ออกได้รวดเร็วจนยาไม่มีโอกาสสัมผัสนานพอที่จะทำให้เกิดการเหนี่ยวนำได้.21
ผลการศึกษาทางคลินิกที่ชัดเจนในเรื่องของการเหนี่ยวนำ expression ของ P-glycoprotein ยังมีไม่มากนัก แต่ก็พอมีตัวอย่างการศึกษาทางคลินิกที่มีผลชัดเจน และเป็นที่ยอมรับในปัจจุบันอยู่บ้าง เช่น การใช้ digoxin, cyclosporine, fexofenadine ซึ่งเป็น substrateของ P-glycoprotein ร่วมกันกับ P-glycoprotein inducer (เช่น rifampicin และ St John's Wort) ในอาสาสมัครสุขภาพดี ซึ่งพบว่ายาในกลุ่มที่เป็น P-glycoprotein inducer มีผลทำให้ค่า bioavailability ของยาดังกล่าวข้างต้นซึ่ง เป็น substrate ของ P-glycoprotein ลดลง และมีระดับยาในเลือดต่ำกว่าช่วงการรักษา (subtherapeutics) เป็นต้น.22-27
Role of ABC Transporters in Cancer Therapy
ปัญหาสำคัญประการหนึ่งในการรักษามะเร็งคือ การดื้อต่อยาเคมีบำบัด ซึ่งมีตั้งแต่การดื้อต่อการรักษามาตรฐานตั้งแต่แรกเริ่มการรักษา เช่น non-small cell cancer, lung cancer และ rectal cancer ซึ่งเรียกว่าเป็น primary resistant หรือ natural resistant แต่ในบางกรณีมะเร็งหลายชนิดที่ตอบสนองดีต่อการรักษาด้วยเคมีบำบัดในช่วงแรกกลับพบว่าเกิดการดื้อต่อยาในระยะต่อมา.
Multidrug resistance (MDR) เป็นปรากฏการณ์ที่เกิดขึ้นหลังจากเซลล์มะเร็งได้สัมผัสกับ ยาต้านมะเร็งเพียงชนิดเดียว แต่สามารถปรับตัวให้ดื้อต่อยาต้านมะเร็งชนิดอื่นๆ ที่มีโครงสร้างและกลไกการออกฤทธิ์ที่แตกต่างจากยาต้านมะเร็งชนิดแรกได้. มีการเปลี่ยนแปลงที่สำคัญเกิดขึ้นในเซลล์มะเร็งเหล่านี้คือ มีการเพิ่มการทำงานของ transmembrane protein ให้ขับสารเคมีออกจากเซลล์, มีการเพิ่มการทำงานของเอนไซม์ในระบบ glutathione detoxification system และมีการเปลี่ยนแปลงยีน และโปรตีนที่เกี่ยวข้องกับกระบวนการ apoptosis กลไกการดื้อเหล่านี้เกี่ยวข้องกับ cell membrane, cytoplasm และ nuclear protein. การเกิด overexpression ของ membrane efflux pumps สัมพันธ์เกี่ยวข้องกับ MDR ซึ่งกลไกการดื้อยาแบบนี้เรียกว่า "typical MDR. หรือ "Classic MDR" 28 ซึ่ง efflux pumps ที่กล่าวถึงก็คือ ABC transporters หรือ ชนิดที่เด่นที่สุดที่มีผลต่อ xenobiotics ต่างๆ และยังรวมไปถึงยาต้านมะเร็งด้วย และมีการศึกษากันมากที่สุดจนถึงปัจจุบันนี้ก็คือ P-glycoprotein ข้อมูลในระยะหลังเริ่มพบว่ามี ABC transporters ชนิดอื่นที่มีบทบาทสำคัญในการดื้อต่อยาเคมีบำบัดในทางคลินิก ได้แก่ MRP1, MRP3 และ BCRP.
ความรู้เกี่ยวกับกลไกการดื้อยาในแบบดังกล่าว ทำให้เราทราบถึงแนวทางในการแก้ปัญหา เช่น การเลี่ยงไปใช้ยาที่ไม่ได้เป็น substrate ของ ABC transporters เช่น alkylating drugs (cyclophosphamide), antimetabolites (5-fluorouracil) และกลุ่ม an- thracycline modified drugs (annamycin และ doxorubicin-peptide) และอีกวิธีหนึ่ง คือการให้ยาที่มีฤทธิ์ยับยั้ง ABC transporter ร่วมไปกับการให้เคมีบำบัด ซึ่งสารที่มีฤทธิ์ดังกล่าวจะช่วยเพิ่มการตอบสนองต่อยาต้านมะเร็ง หรือที่เราเรียกว่าเป็น "Chemosensitizers".
หลังจากที่ Juliano และ Ling ได้รายงานเกี่ยวกับ Phosphoglycoprotein ที่ express ใน drug- resistant Chinese hamster ovary cells เป็นครั้งแรกในปี พ.ศ. 251928 และนำไปสู่การค้นพบ P-glycoprotein ซึ่งเป็น ATP-transporter จนในปี พ.ศ. 2524 verapamil เป็นยาตัวแรกที่มีรายงานว่ามีฤทธิ์เป็น chemosensitizer.29 จากนั้นได้มามีความพยายามในการพัฒนายาที่จะเป็น chemosensitizer เรื่อยมา ปัจจุบันเราสามารถแบ่งยากลุ่มนี้เป็น 3 generations ตามคุณสมบัติและผลไม่พึงประสงค์ จากยาดังกล่าว คือ
กลุ่ม First-generation agents ได้แก่ Verapamil, cyclosporine, amiodarone, และ tamoxifen พบว่ามีฤทธิ์ยับยั้ง P-glycoprotein แต่ยากลุ่มนี้ไม่สามารถนำไปใช้ทางคลินิกได้ เนื่องจาก ขนาดที่ยาที่จะมีฤทธิ์ในการเป็น chemosensitizer จำเป็นต้องใช้ในขนาดสูงจนทำให้เกิดอาการไม่พึงประสงค์จากยาเหล่านี้ เช่น การเกิดพิษต่อหัวใจของ verapamil ซึ่งต่อมาได้มีการพยายามพัฒนาเป็น (R)-verapamil และอนุพันธ์ที่มีพิษต่อหัวใจน้อยกว่า (S)-verapamil มาใช้. นอกจากนี้ ความล้มเหลวในการศึกษาอาจเกิดจากในระยะนั้นยังมีข้อมูล in vivo อยู่ไม่มากพอ และไม่ได้พิจารณาในแง่มุมของ P-glycoprotein expression ใน tumors ซึ่งมีความเปลี่ยนแปลงและแตกต่างกัน ทำให้ล้มเหลวใน cli-nical trial phase III ซึ่งพบว่า cyclosporine และ verapamil ไม่สามารถทำให้ผลการรักษาดีขึ้นได้.
Second-generation agents เป็นอนุพันธ์ของยากลุ่มแรกที่มีฤทธิ์สูงกว่าและมีพิษน้อยกว่ายาที่มีการศึกษาได้แก่ PSC 833 (valspodar) ซึ่งเป็นอนุพันธ์ของ cyclosporine D มีฤทธิ์สูงกว่า cyclosporin A 10 เท่า และไม่มีฤทธิ์กดภูมิคุ้มกัน, VX-710 (biricodar) ซึ่งมีฤทธิ์ต่อทั้ง MDR1 และ MRP1 ใน in vitro study ซึ่งอาจมี drug interaction profile แตกต่างจาก PSC 833 แต่ยากลุ่มนี้ก็ยังพบว่ามี drug interaction โดยเฉพาะอย่างยิ่งกับ CYP3A4 ซึ่งอาจรบกวน metabolism ของยาต้านมะเร็งบางชนิด เช่น paclitaxel และยังพบว่ามีผลทำให้เกิดอาการไม่พึงประสงค์คือ ataxia.
Third-generation agents ซึ่งพยายามพัฒนาให้เป็นยาไม่มีผลต่อ pharmacokinetics interaction ในปัจจุบันมียาที่ได้ผ่านการศึกษามา จนถึงขั้นที่กำลังศึกษาผลทางคลินิก (Phase III) อยู่หลายชนิด เช่น LY33597 (Zosuquidar), XR 9576 (Tariquidar), R101933 (Laniquidar) และ ONT0093 เป็นต้น ซึ่งยังต้องใช้เวลาเพื่อรอผลการศึกษาถึงประสิทธิภาพ ในการเสริมการรักษามะเร็งที่ดื้อต่อเคมีบำบัด และติดตามเรื่องความปลอดภัยของยาต่อไป.30,31
ข้อมูล ณ ปัจจุบัน จาก randomized trials ยังมีผลการศึกษาไม่มากนัก ที่สามารถชี้ให้เห็นว่าการใช้ P-glycoprotein inhibitor ร่วมกับ ยาต้านมะเร็ง สามารถเพิ่ม overall survival ได้อย่างมีนัยสำคัญทางสถิติเมื่อเทียบกับกลุ่มที่ไม่ได้รับ P-glycoportein inhibitor ตามทฤษฎีที่คาดการณ์กันไว้ ทั้งนี้ อาจเป็น จากกรณีที่มีการคัดเลือกผู้ป่วยเข้าร่วมในการศึกษาและการออกแบบการศึกษาที่ไม่เหมาะสม ทำให้ผลการศึกษาที่ออกมาได้ผลในเชิงลบ เนื่องจากมีปัจจัยอีกหลายอย่างที่ต้องพิจารณาร่วม เช่น โรคหรือชนิดของ มะเร็ง ซึ่งต้องพิจารณาว่า Protein transporter หลัก ที่ express ในมะเร็งชนิดนั้นเป็นชนิดไหน อย่างไร กลุ่มประชากร และเชื้อชาติก็อาจมีผลจาก polymorphisms มาเกี่ยวข้องร่วมด้วย. นอกจากนี้ ชนิดของยาต้านมะเร็งที่ศึกษาหากมีการนำผู้ป่วยที่ใช้ยา combination ที่มียาที่ไม่ได้เป็น substrate ของ P-glycoprotein เป็นหลักมาศึกษา เช่น Cisplatin หรือ cytosine arabinoside (Ara-C) ก็อาจจะไม่สามารถแสดงผลที่เกี่ยวข้องกับสมมติฐานที่ตั้งไว้.
แม้ว่าการศึกษาเกี่ยวกับ ABC transporters ได้ดำเนินการศึกษากันมาอย่างต่อเนื่องมากว่า 30 ปี แต่ผลการศึกษาส่วนใหญ่ที่ได้มาแม้ว่าสามารถอธิบาย เหตุผลบางส่วนในเชิงลึกและซับซ้อนได้ แต่ก็เป็นข้อมูลจากการศึกษาแบบ in vitro และ in vivo โดยยังคงเป็นการศึกษาในสัตว์ทดลอง. การศึกษาในทางคลินิกยังมีน้อยและมีข้อจำกัดในการศึกษามาก และจำเป็นต้องใช้เวลานานในการติดตามผลการศึกษา และเนื่องจาก ABC transporters มีความสัมพันธ์ กับปัจจัยในระบบต่างๆ ของร่างกายมากมายและเป็นไปอย่างซับซ้อน ซึ่งยากต่อการแปลผลอีกทั้งยังต้องการ ข้อมูลความจำเพาะ และความแตกต่างในหลายๆ ด้านที่เชื่อมโยงกันเพื่อที่จะอนุมานผลการศึกษาจาก in vitro และการศึกษาในสัตว์ทดลองมายังผลที่คาดว่าจะเกิดขึ้นในมนุษย์ แต่อย่างน้อยผลการศึกษาทางคลินิกบางส่วน ก็ยังเป็นข้อมูลที่สำคัญและเป็นประโยชน์ ซึ่งสามารถนำมาใช้เพื่อเฝ้าระวังและป้องกันอาการ ไม่พึงประสงค์จากการใช้ยา และอาจช่วยคาดคะเน ผลกระทบต่อประสิทธิผลของยาที่เกิดจากปฏิกิริยาระหว่างกันของยาได้.
เอกสารอ้างอิง
1. Wilkinson G. Pharmacokinetics: the dynamics of drug absorption, distribution, and elimination. In : Hardman JG, Limbird LE, Gilman AG, editors. Goodman & Gilman’s the pharmacological basis of therapeutics. New York : McGraw-Hill; 2001. p. 3-29.
2. Hagenbuch B, Meier PJ. The superfamily of organic anion transporting polypeptides. Bio-chim Biophys Acta 2003; 1609:1-18.
3. Schinkel AH, Jonker JW. Mamlian drug efflux transporters of the ATP binding cassette (ABC) family : and overview. Adv Drug Deliv Rev 2003; 55:3-29.
4. Marzolini C, Paus E, Bulcin T, Kim RB. Polymorphisms in human MDR1 (P-glycoprotein) : recent advances and clinical relevance. Clin Pharmacol Ther 2004; 75:13-33.
5. Ameyaw M-M, Regateiro F, Li T, et al. MDR1 pharmacogenetics : frequency of the C3435T mutation in exon 26 is significantly influenced by ethnicity. Pharmacogenetics 2001; 11:217-21.
6. Elmore JG, Moceri VM, Carter D, et al. Breast carcinoma tumor characteristics in black and white women. Cancer 1998; 83:2509-15.
7. Thiebaut F, Tsuruo T, Hamada H, et al. Cellular localization of the multidrug resistance gene product P-glycoprotein in normal human tissues. Proc Natl Acad Sci USA 1987; 84: 7735-8.
8. Cordon-Cardo C, O'Brien JP, Boccia J, et al. Expression of the multidrug resistance gene product (P-glycoprotein) in human normal and tumor tissues. J Histochem Cytochem1990; 38:1277-87.
9. Sparreboom A, van Asperen J, Mayer U, Schinkel AH, Smit JW, Meijer DK, et al. Limited oral bioavailability and active epithelial excretion of paclitaxel (Taxol) caused by P-glycoprotein in the intestine. Proc Natl Acad Sci USA 1997; 94:2031-5.
10. Schinkel AH, Wagenaar E, van Deemter L, Mol CA, Borst P. Absence of the mdr1a P-Glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J Clin Invest 1995; 96:1698-705.
11. Kim RB, Fromm MF, Wandel C, Leake B, Wood AJ, Roden DM, et al. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors.J Clin Invest 1998; 101:289-94.
12. Lown KS, Mayo RR, Leichtman AB, Hsiao HL, Turgeon DK, Schmiedlin-Ren P, et al. Role of intestinal P-glycoprotein (mdr1) in interpatient variation in the oral bioavailability of cyclo-sporine. Clin Pharmacol Ther 1997; 62:248-60.
13. Drescher S, Glaeser H, Murdter T, Hitzl M, Eichelbaum M, Fromm MF. P-glycoprotein-mediated intestinal and biliary digoxin transport in humans. Clin Pharmacol Ther 2003; 73:223-31.
14. Burton PS, Conradi RA, Hilgers AR, et al. Evidence for a polarized efflux system for peptides in the apical membrane of Caco-2 cells. Biochem Biophys Res Commun 1993; 190:760-6.
15. Masuda M, I'izuka Y, Yamazaki M, Nishigaki R, Kato Y, Niinuma K, et al. Methotrexate is excreted into the bile by canalicular multispecific organic anion transporter in rats. Cancer Res 1997; 57:3506-10.
16. Chu XY, Kato Y, Sugiyama Y. Multiplicity of biliary excretion mechanisms for irinotecan, CPT-11, and its metabolites in rats. Cancer Res 1997; 57:1934-8.
17. Sasaki M, Suzuki H, Ito K, Abe T, Sugiyama Y. Transcellular transport of organic anions across a double-transfected Madin-Darby canine kidney II cell monolayer expressing both human organic anion-transporting polypeptide (OATP2/ SLC21A6) and multidrug resistance-associated protein 2 (MRP2/ABCC2). J Biol Chem 2002; 277:6497-503.
18. You G. Structure, function, and regulation of renal organic anion transporters. Med Res Rev 2002; 22:602-16.
19. Shitara Y, Horie T, Sugiyama. Transpoters a determinant of drug clearance and tissue distribution. Eur J Pharm Sci 2006; 27:425-46.
20. Durr D, Stieger B, Kullak-Ublick GA, et al. St. John's Wort induces intestinal P-glycoprotein/MDR1 and intestinal and hepatic CYP3A4. Clin Pharmacol Ther 2000; 68:598-604.
21. Jette L, Beaulieu E, Leclerc J-M, et al. Cyclosporin A treatment induces overexpression of P-glycoprotein in the kidney and other tissues. Am J Physiol 1996; 270:F756-65.
22. Greiner B, Eichelbaum M, Fritz P, et al. The role of intestinal P-glycoprotein in the interaction of digoxin and rifampin. J Clin Invest 1999; 104:147-53.
23. Hamman MA, Bruce MA, Haehner-Daniels BD, et al. The effect of rifampin administration on the disposition of fexofenadine. Clin Pharmacol Ther 2001; 69:114-21.
24. Lippert C, Ling J, Brown P, et al. Mass balance and pharmacokinetics of MDL16,455A in healthy male volunteers [abstract]. Pharm Res 1999; 12:S390.
25. Cvetkovic M, Leake B, Fromm MF, et al. OATP and P-glycoprotein transporters mediate the cellular uptake and excretion of fexofenadine. Drug Metab Dispos 1999; 27:866-71.
26. Hebert MF, Roberts JP, Prueksaritanont T, et al. Bioavailability of cyclosporine with concomitant rifampin administration is markedly less than predicted by hepatic enzyme induction. Clin Pharmacol Ther 1992; 52:453-7.
27. Wacher VJ, Silverman JA, Zhang Y, et al. Role of P-glycoprotein and cytochrome P450 3A in limiting oral absorption of peptides and peptidomimetics. J Pharm Sci 1998; 87:1322-30.
28. Juliano RL, Ling V. A surface glycoprotein modulating drug permeability in Chinese hamster ovary cell mutants. Biochim Biophys Acta 1976; 455:152-62.
29. Tsuruo T, Iida H, Tsukagoshi S, Sakurai Y: Overcoming of vincristine resistance in P388 leukemia in vivo and in vitro through enhanced cytotoxicity of vincristine and vinblastine by verapamil. Cancer Res 1981; 41:1967-72.
30. Thomas H, Coley HM. Overcoming Multidrug Resistance in Cancer : An Update on the Clinical Strategy of Inhibiting P-Glycoprotein. Cancer control 2003; 10(2):159-65.
31. Leonard GD, Fojo T, Bates SE. The role of ABC transporter in clinical practice. The Oncologist 2003; 8:411-24.
32. Baer MR, George SL, Dodge RK, et al. Phase 3 study of the multidrug resistance modulator PSC-833 in previously untreated patients 60 years of age and older with acute myeloid leukemia : Cancer and Leukemia Group B Study 9720. Blood 2002; 100:1224-32.
33. Choi CH. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int 2005; 5:30.
34. Loscher W, Potschka H. Role of drug efflux transports in the brain for drug disposition and treatment of brain diseases. Prog Neurobiol 2005; 76:22-76.
35. Takano M, Yumoto R, Murakami T. Expression and function of efflux drug transporters in the intestine. Pharmacol Ther 2006; 109: 137-61.
เด่นพงศ์ พัฒนเศรษฐานนท์, Ph.D., ภ.บ., อาจารย์
สาขาเภสัชกรรมปฏิบัติ คณะเภสัชศาสตร์ มหาวิทยาลัยขอนแก่น
- อ่าน 19,601 ครั้ง
 พิมพ์หน้านี้
พิมพ์หน้านี้